Microsoft Word can do a lot more things for you than you can actually know! Most people use MS-Word for very basic things. But at times you may face situations where you feel that MS-Word will not be able to help you and you will have to do it all manually. For example, what if you need to find and replace all the images in a Word document? Do you think MS-Word can do this? Well, yes! A reader of TechWelkin sent me a query. She said that she is a writer and is working on a large manuscript in MS-Word. She used a tiny image as a separator for various chapters and sections in the document. Now she wants to replace that image with a new one. But there are hundreds of copies of the image. So, will she have to replace each image one by one? Let’s explore the answer!
MS-Word can help in the above given scenario. There is an automatic way to find all the images in a document and then replace them all with a new image. Please note that this method will find all the images and it will replace all of them with a new one. With this method, we can not be selective about images. Here are the steps:
- Open the MS-Word document in which replacement is to be done.
- Insert the new image at the top of the document.
- Select the newly inserted image and press Ctrl + C to copy it.
- Now delete the newly inserted image.
- Press Ctrl + H to open the Find and Replace box.
- Put ^g in the Find what box and ^c in the Replace with box
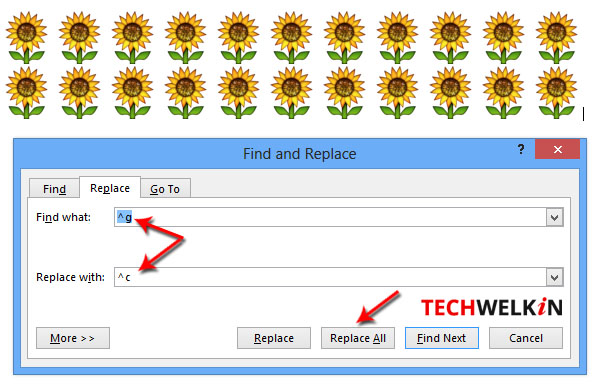
Method of finding all images and replacing them with a new one.
Now press Replace All button to see the magic!
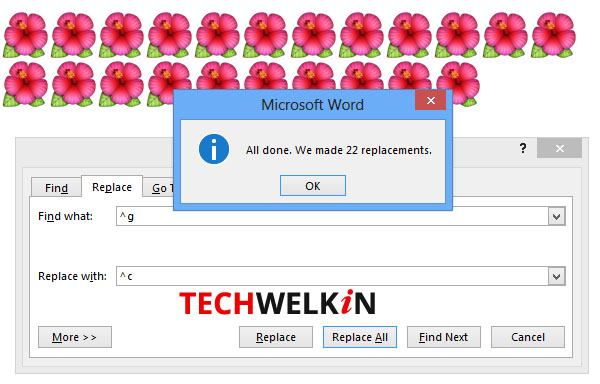
All images have been replaced in MS Word.
You will see that all the images in the document will be replaced with the new image. Of course, the replacement operation will also replace the images that you did not want to replace. In such a case you may need to re-insert the images that were not to be replaced. This method is useful in a scenario where:
- the document contains only those images that are to be replaced
- the document contains a large number of images that need to be replaced and a smaller number of images that should not be replaced
The first scenario is straightforward. In the second scenario, you’ll be doing much less manual work by re-inserting images that were replaced as a side-effect.
The method works by replacing all graphics (denoted by ^g) with the clipboard content (denoted by ^c). Therefore, essentially, you can replace all images with anything that you can copy to clipboard.
To keep your document’s pagination intact, you should use the same size new image.
I hope this MS-Word tip was useful for you. If you have any question on this topic, please feel free to ask me through comments. I will try to assist you. Thank you for using TechWelkin.
How can u replace multiple pictures with other multiple pictures in Microsoft word
And if they are in a table how can i select them all and replace them with others
Thanks in advance
is there any way to replace very tiny image ocurring muliple times in the doc, actually i have a solved exam paper downloaded from portal so it has cross n correct tick against all the options with red n green colour respectively, any way to delete them without tempering the questions and options..
NOTE.- Options are also in image format(looks like scanned one).
where is the select command found?
Hello – thanks for posting this advice. I know this is a little old, but I’m hoping you can assist anyway. I can’t seem to accomplish this for graphics that are inside of headers. “Find and Replace” can’t find the graphics in the headers. Thanks!
Awesome article. I didn’t imagine that it was possible!!!!
My Image Is Not Getting Replaced It Says Word Has Finished Searching And Has Replaced 0 Images
Same problem with me. I am using Office 2016 with Windows 10.
I have the same problem. Also when I try and press ctrl g I get nothing.
I have to use VBA for Shapes. Find ^g not yet capable even for Word 365.
Hi all,
I want to delete all text in my ms word without deleting image. Is that possible. If so help me to resolve those things.
many thanks in advance
Chandramouli.S
Type ^? in Find what box and leave the Replace with box blank. Click Replace All button. This will delete all the text but leave images in Word document.
hi, is there a way of replacing specific images (not all) in MS words, i.e. not having to replace them one by one of course. the thing is i have multiple images in my file and I just want to replace one image that has been used several times on the document.
Thanks.
In MS-Word, it is not possible to replace images on the basis of them being duplicates.
This was really helpful. Thanks!
Is there an easy way to replace a word, say ‘Website’ with an icon (a jpg format)?
Hi Richard, yes, there is an easy way! And to explain it we have just published a new article on this topic. Learn how to replace text with an image in MS Word.
Dear Page Admin
When I replace an image the size does not fit. Please help me.
Hello Abdul, in this method of replacing images in MS Word, you can not specify image size. So, you should try to make sure that the sizes of both images is same.